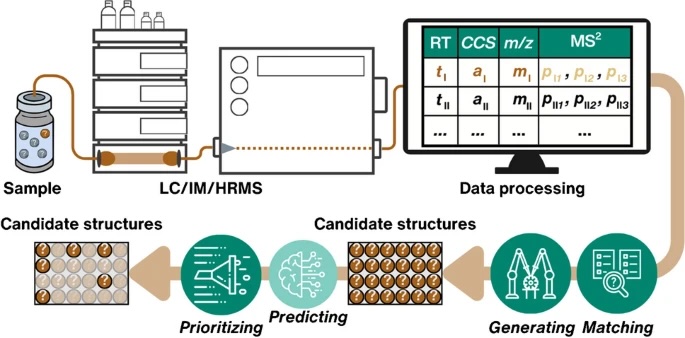
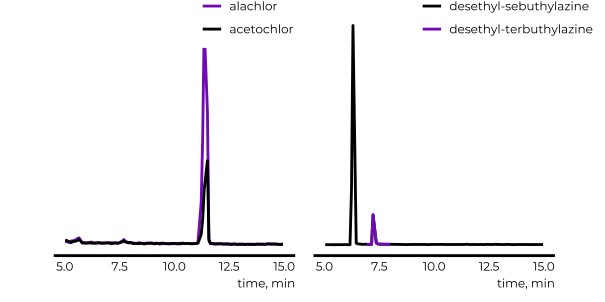
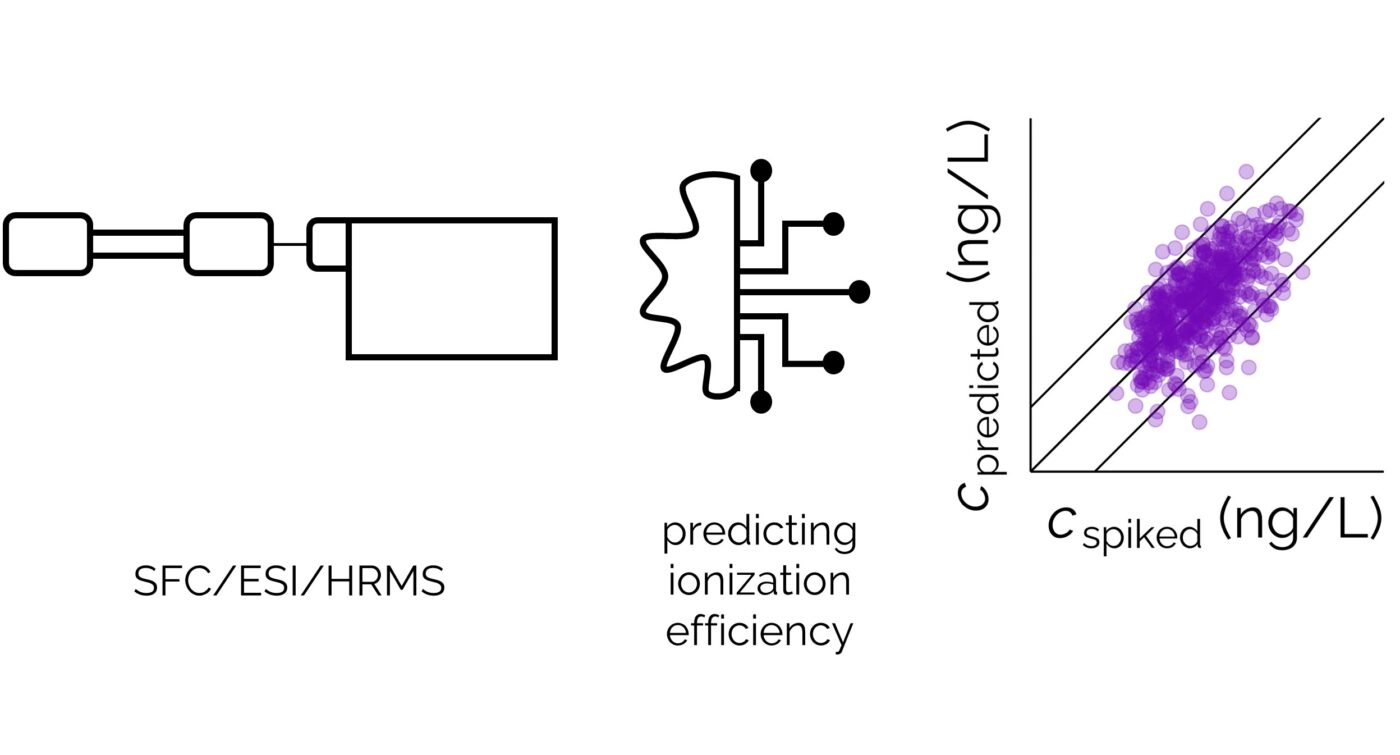
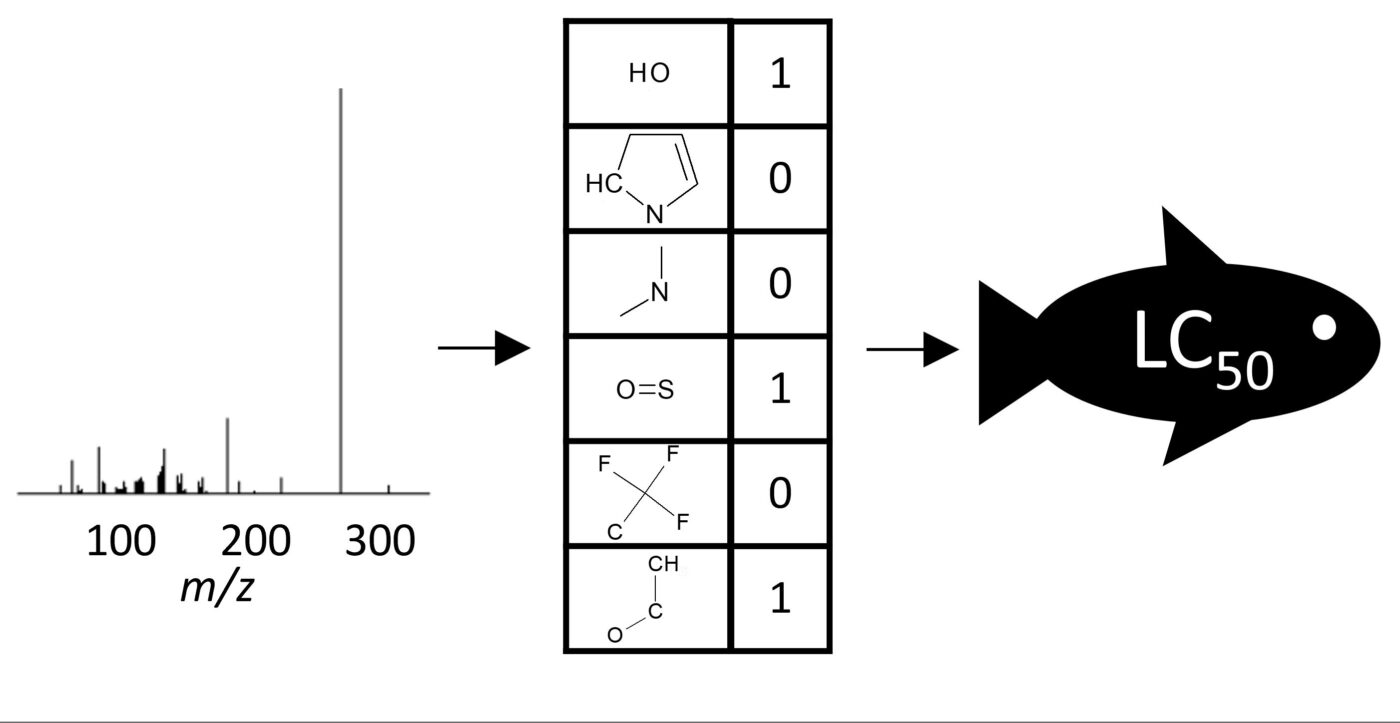
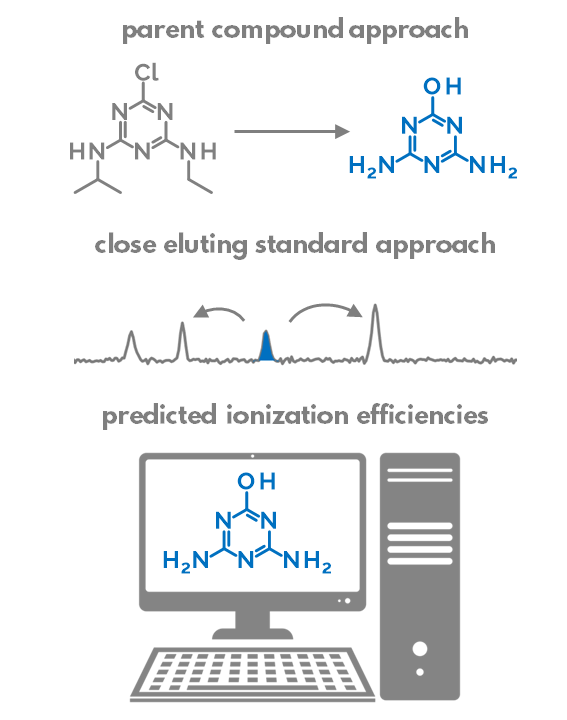
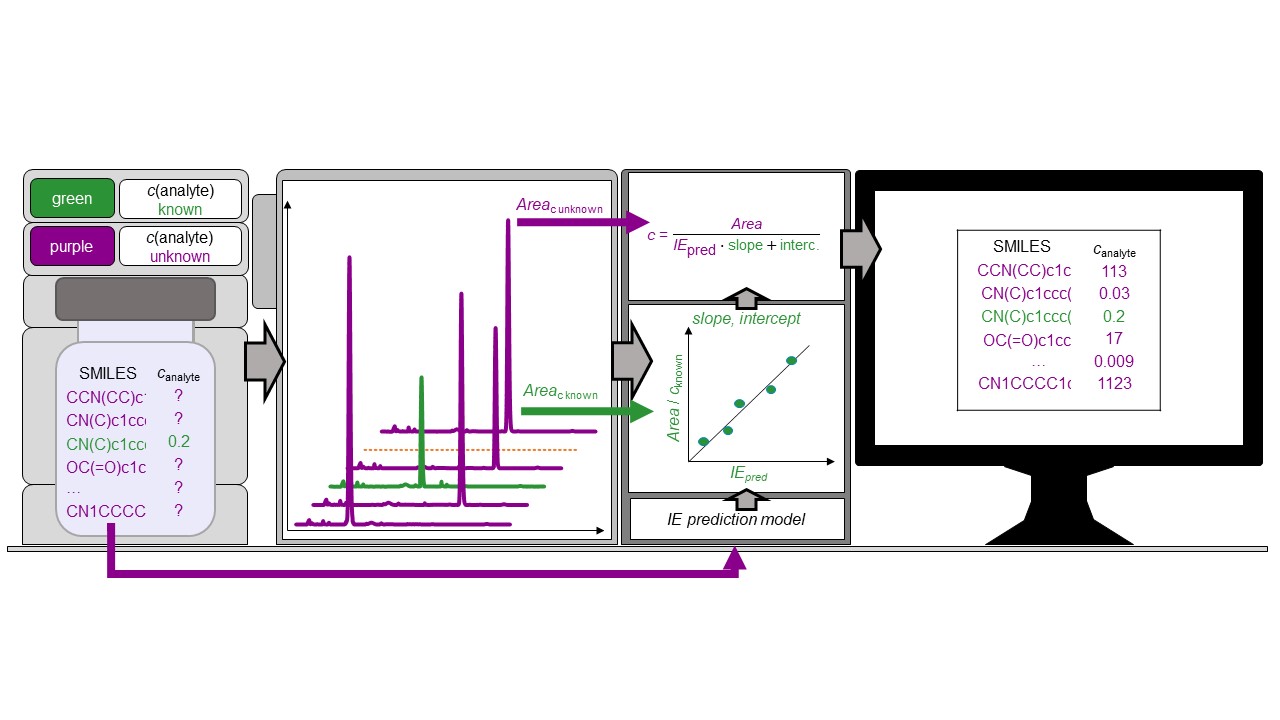
Do experimental projection methods outcompete retention time prediction models in non-target screening? A case study on LC/HRMS interlaboratory comparison data
Louise Malm, Anneli Kruve
Analyst 2025
DOI: 10.1039/d5an00323g
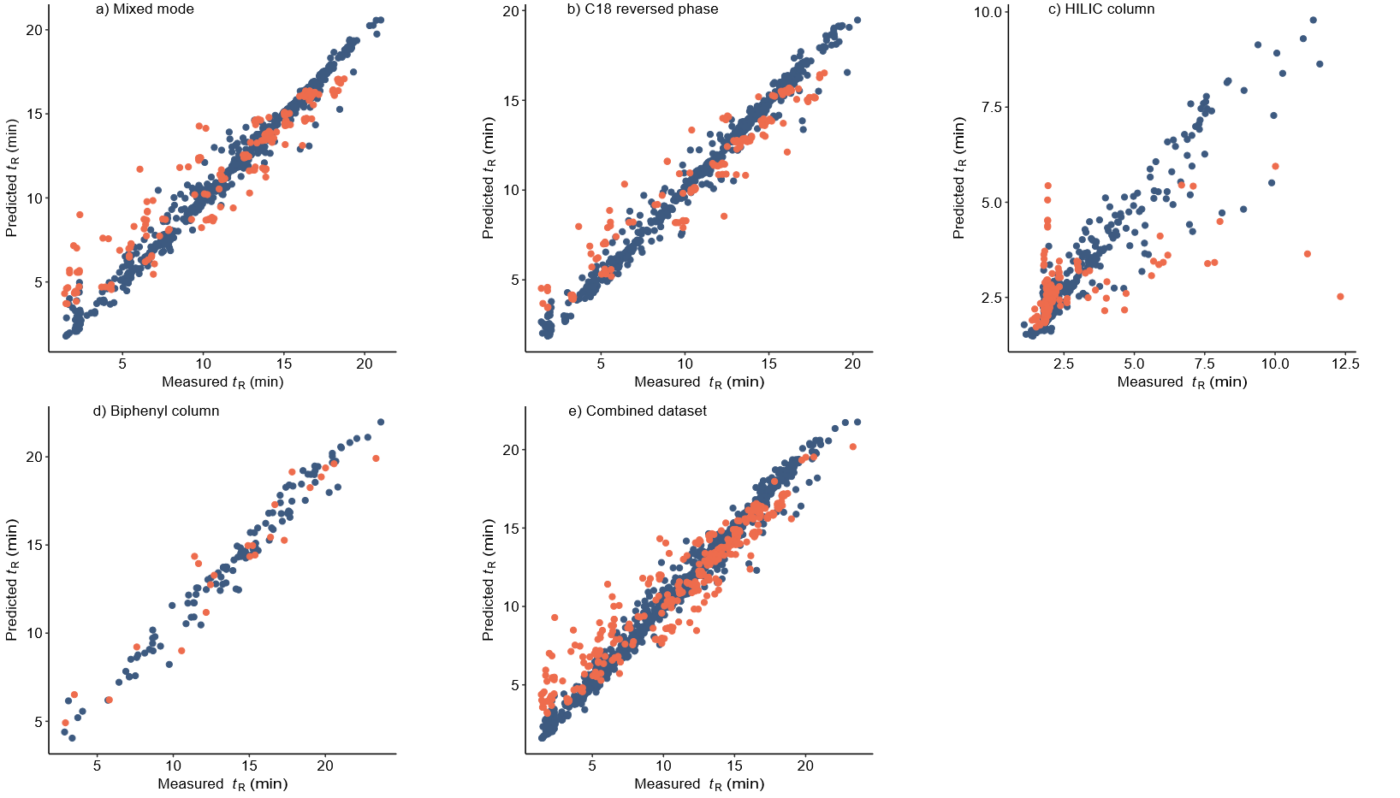
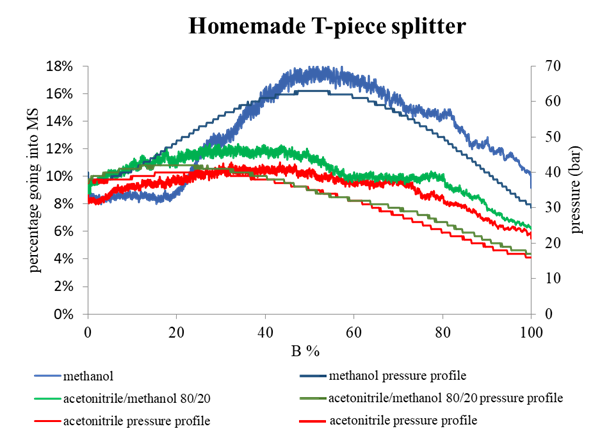
Retention time (RT) is essential in evaluating the likelihood of candidate structures in nontarget screening (NTS) with liquid chromatography high resolution mass spectrometry (LC/HRMS). Approaches for estimating RT of candidate structure can broadly be divided into projection and prediction methods. The first approach takes advantage of public databases of RTs measured on similar chromatographic systems (CSsource) and projects these to the chromatographic system applied in the NTS (CSNTS) based on a small set of commonly analyzed chemicals. The second approach leverages machine learning (ML) model(s) trained on publicly available retention time data measured on one or more chromatographic systems (CStraining). Nevertheless, the CSsource and CStraining might differ substantially from CSNTS. Therefore, it is of interest to evaluate the generalizability of projection models and prediction models in CSs routinely applied in NTS.
Here we take advantage of the recent NORMAN interlaboratory comparison where 41 known calibration chemicals and 45 suspects were analyzed to evaluate both the projection and prediction approach on 37 CSs. The accuracy of both approaches was directly linked to the similarity of the CS and the pH of the mobile phase as well as the column chemistry were found to be most impactful. Furthermore, for cases where CSsource and CSNTS differ substantially but CStraining and CSNTS were similar, prediction models often performed on bar with the projection models. These findings highlight the need in accounting for the mobile phase and column chemistry in ML model training and selecting the prediction model for RT.

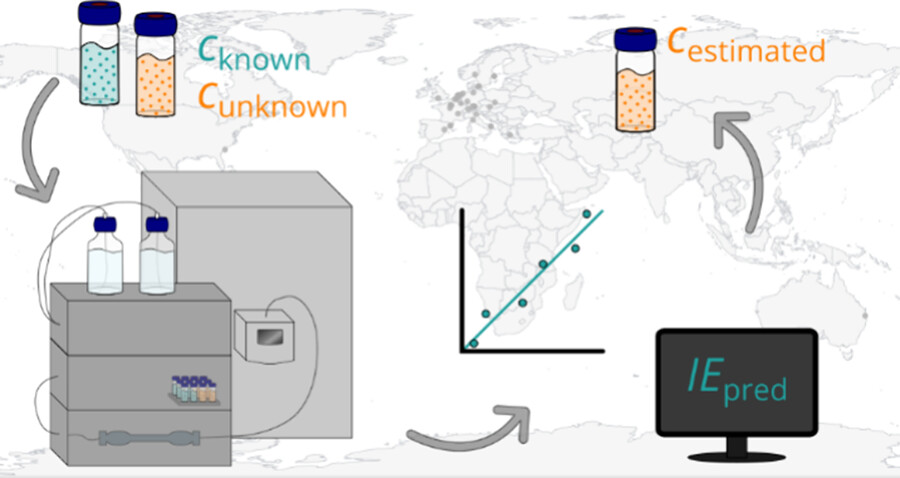
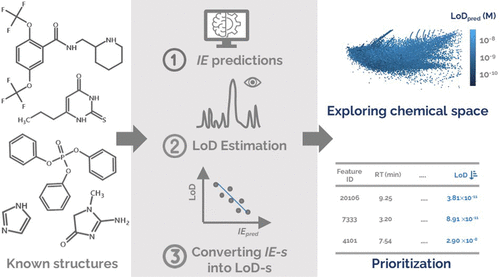
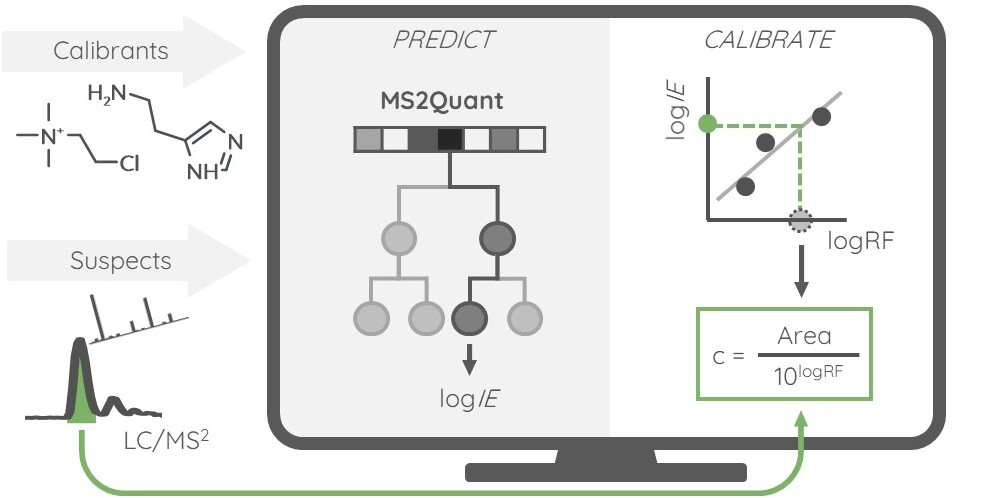
Active Learning Improves Ionization Efficiency Predictions and Quantification in Nontargeted LC/HRMS
Wei-Chieh Wang, Nahid Amini, Carolin Huber, Meelis Kull, Anneli Kruve
Anal. Chem. 2025
DOI: 10.1021/acs.analchem.5c00816
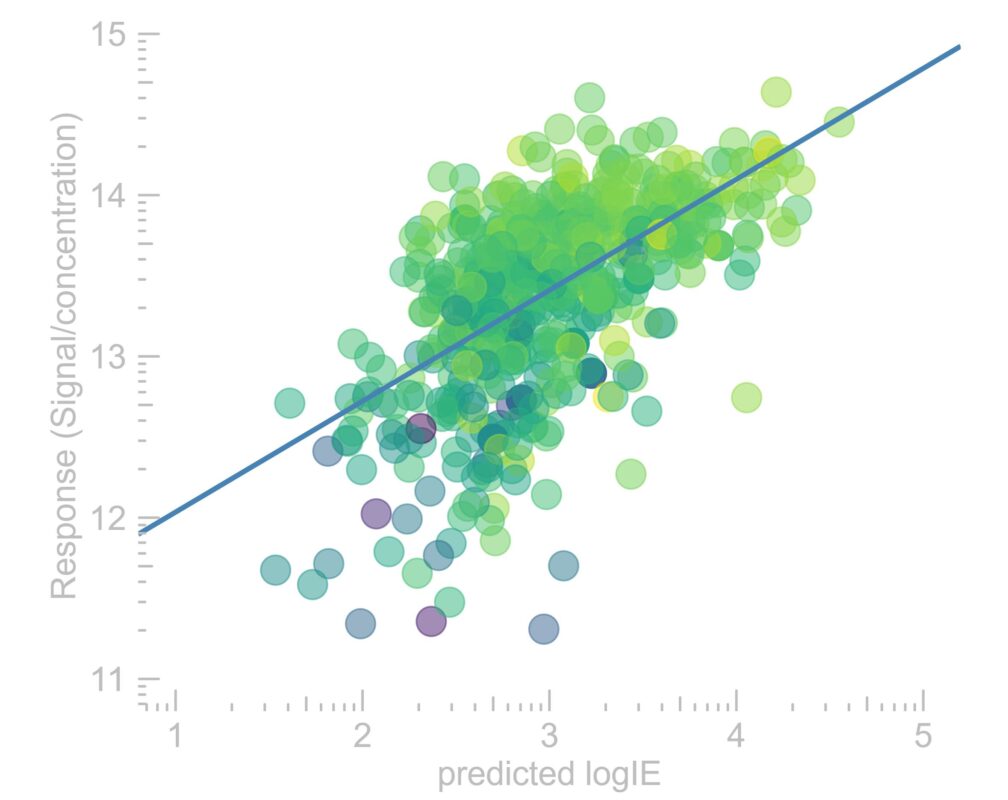
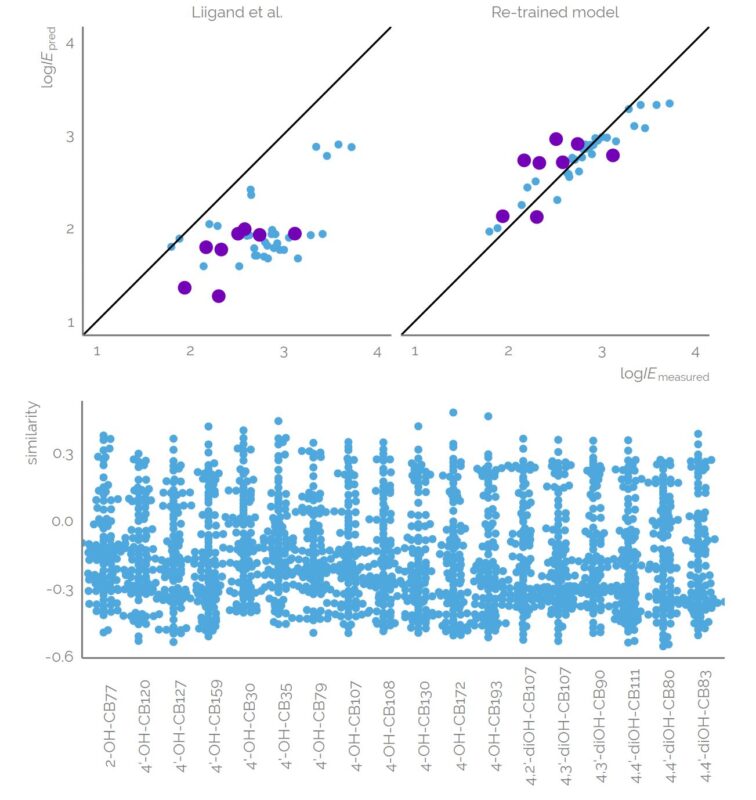
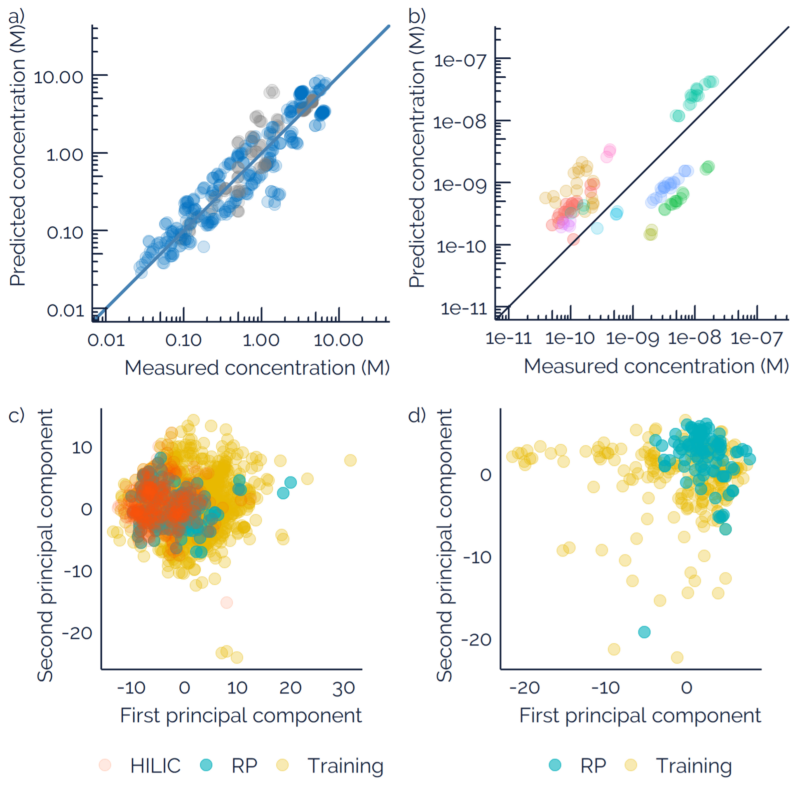
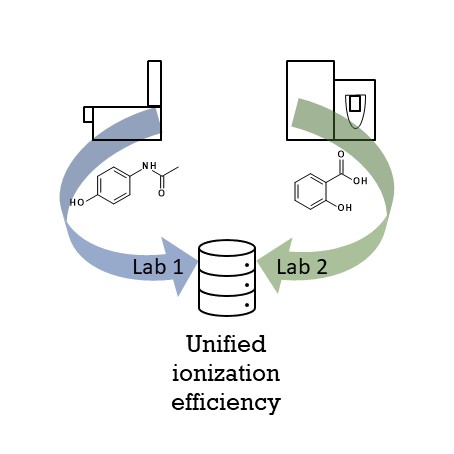
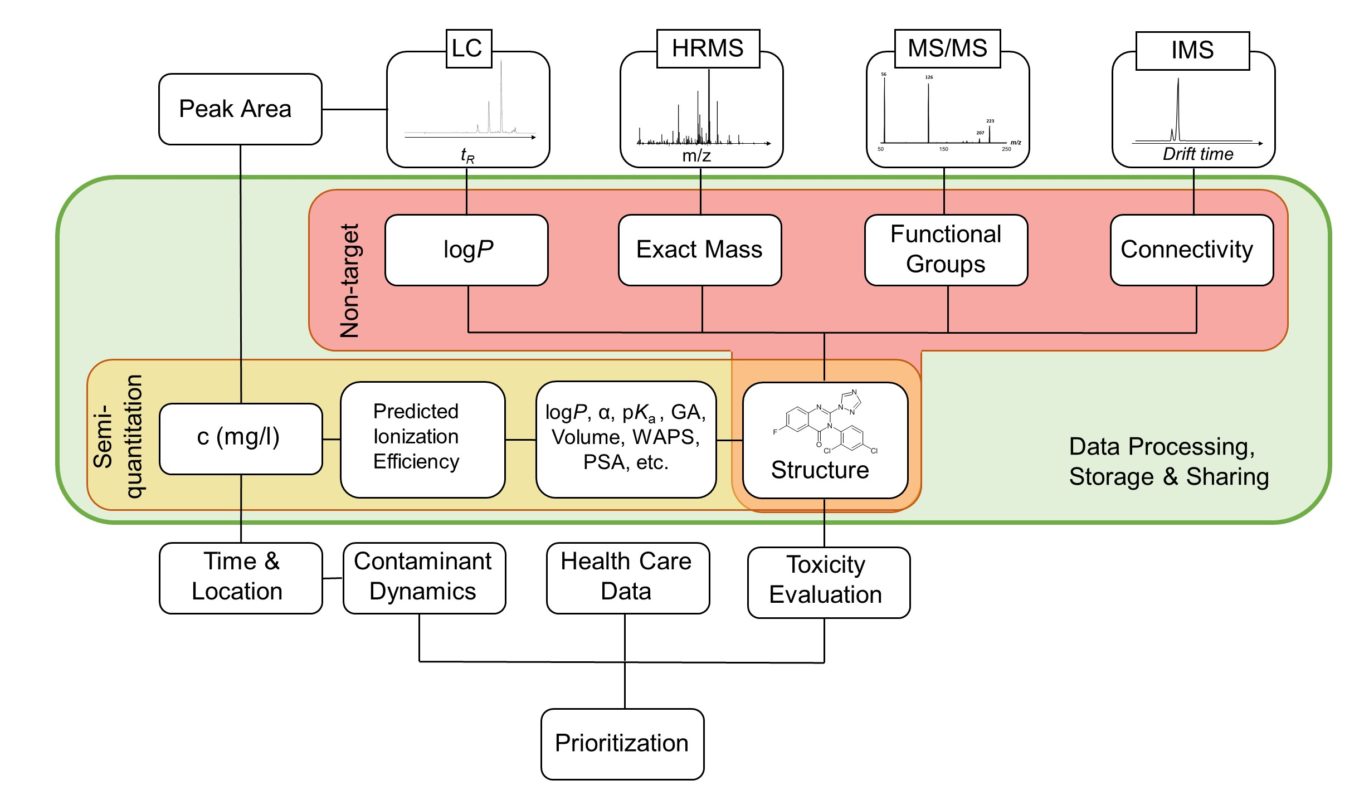
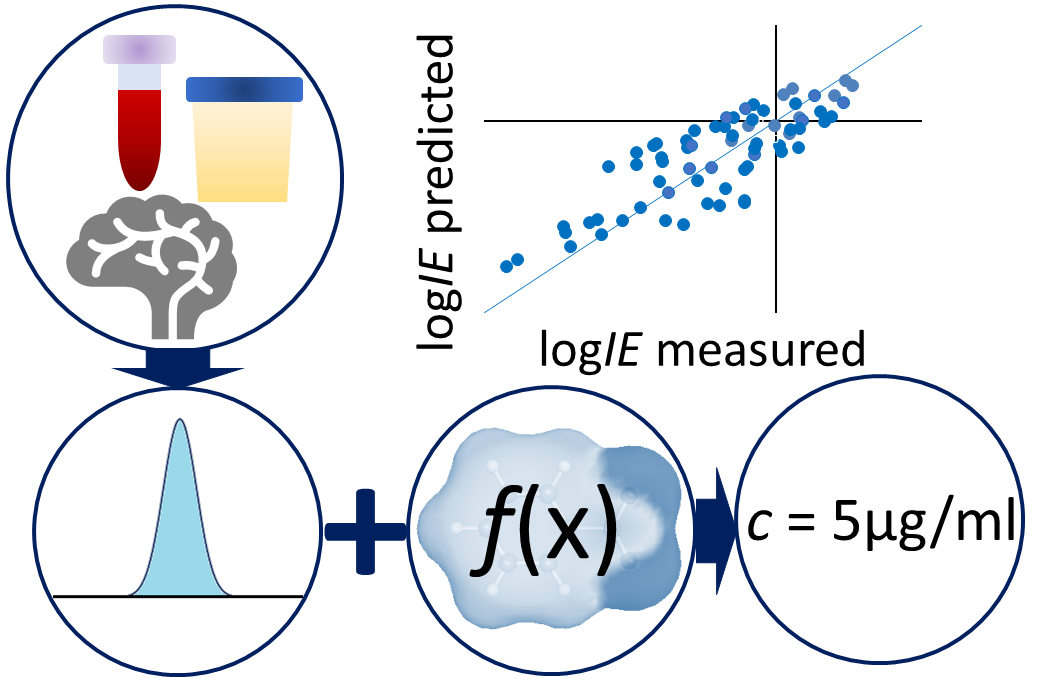
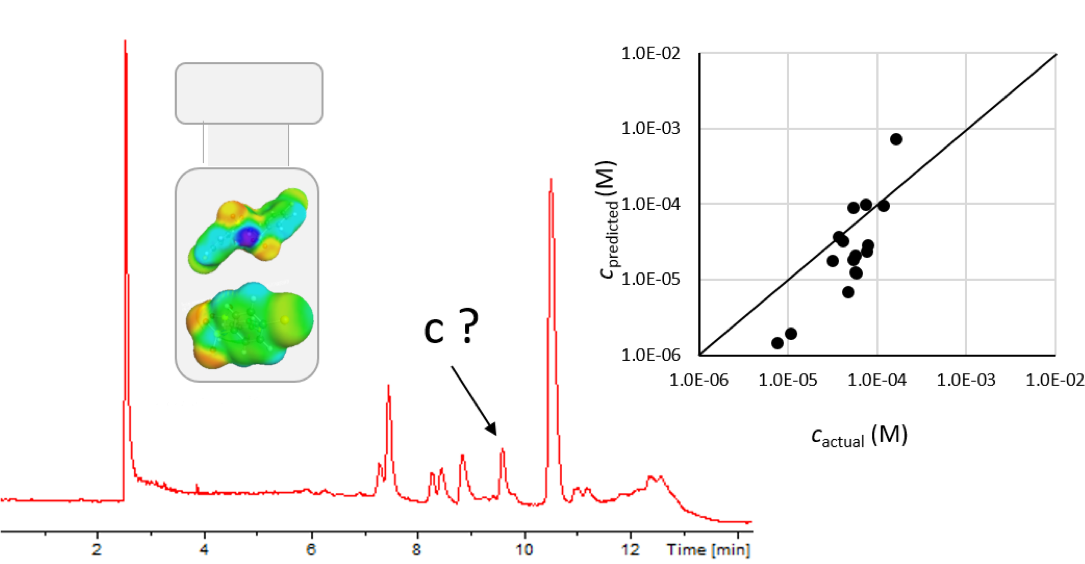
Liquid chromatography electrospray ionization high-resolution mass spectrometry (LC/ESI/HRMS) is frequently employed in nontargeted screening (NTS) due to its high selectivity and sensitivity. However, data interpretation is challenging since the number of chemical standards available for quantification is limited and the response of the chemicals vastly differs depending on their structure and analysis conditions. Therefore, machine learning (ML) models have been utilized to predict ionization efficiency (IE) and enable the quantification of detected chemicals. It has been observed that the error in the predictions is high for chemicals structurally different from the training data. To enlarge the training set and to accurately predict the IE given a limited labeling budget, active learning (AL) is proposed to acquire informative data points from the targeted chemical space. In the current study, four AL approaches (clustering-based, uncertainty-based, mix, and anticlustering) and a baseline approach (random) were evaluated for IE prediction. The RMSE of the IE in the targeted space dropped significantly (up to 0.3 log units) after a single AL iteration, highlighting the necessity of chemical space exploration before ML model execution. Clustering-based AL reduced the RMSE least, while the uncertainty-based AL was inefficient if ten or more chemicals were sampled in one iteration, thereby reducing its practicality. Finally, expanding the chemical space improved the quantification accuracy from a fold error of 4.13× to 2.94× for five natural products in Alpinia officinarum, thereby demonstrating the need for updating the chemical space coverage of the training set.

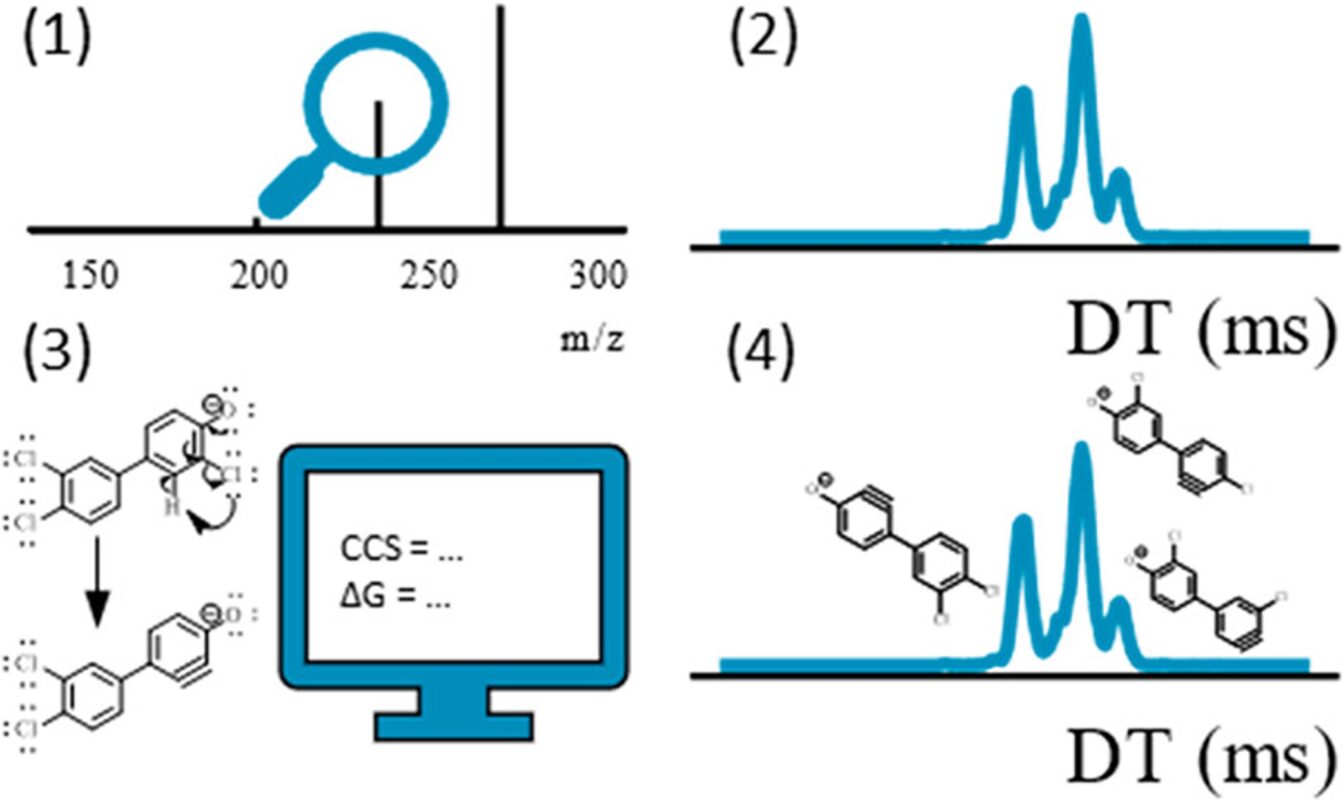
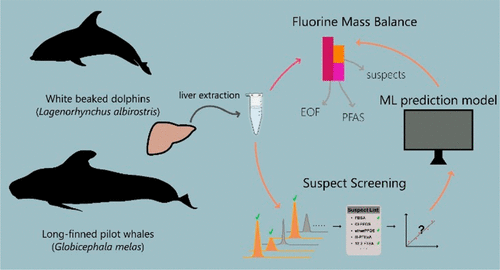
Examining environmental matrix effects on quantitative non-targeted analysis estimates of per-and polyfluoroalkyl substances
Shirley Pu, James P McCord, Rebecca A Dickman, Nickolas A Sayresmith, Helen Sepman, Anneli Kruve, Diana S Aga, Jon R Sobus
Anal Bioanal Chem 2025
DOI: 10.1007/s00216-025-05796-1
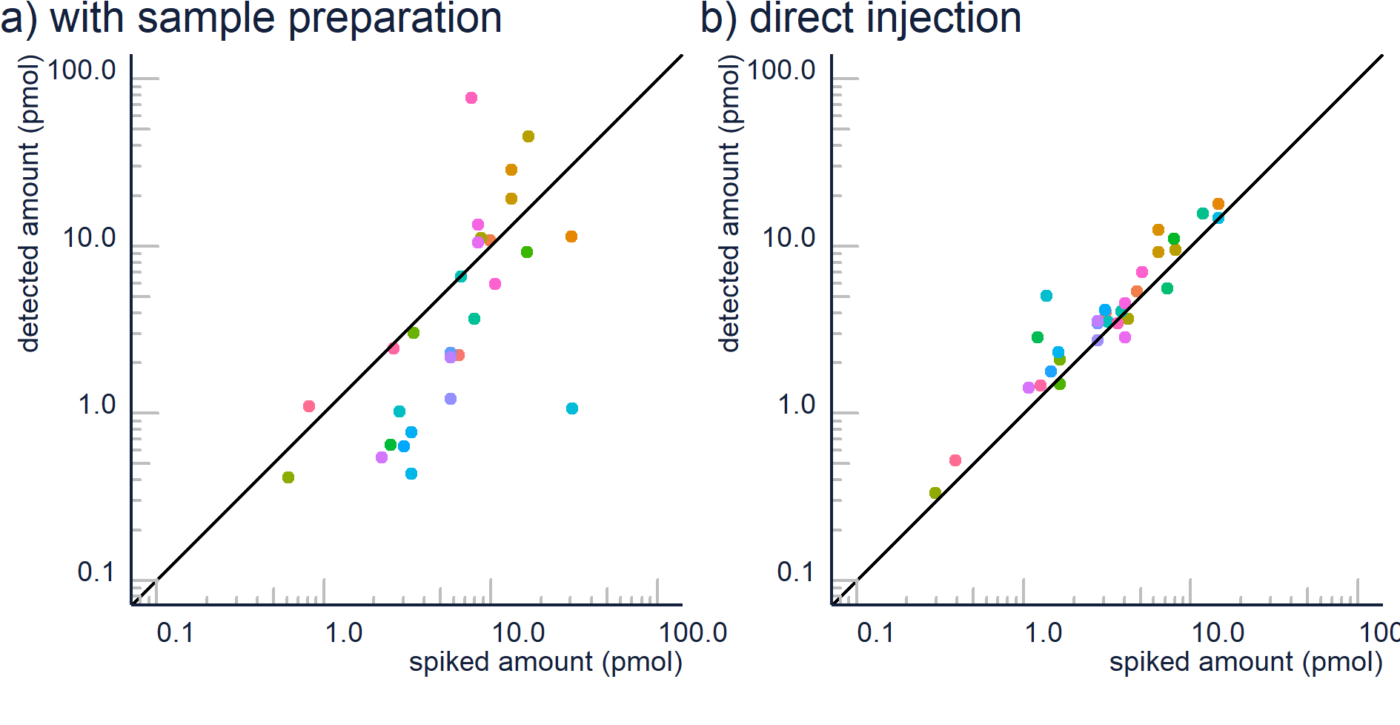
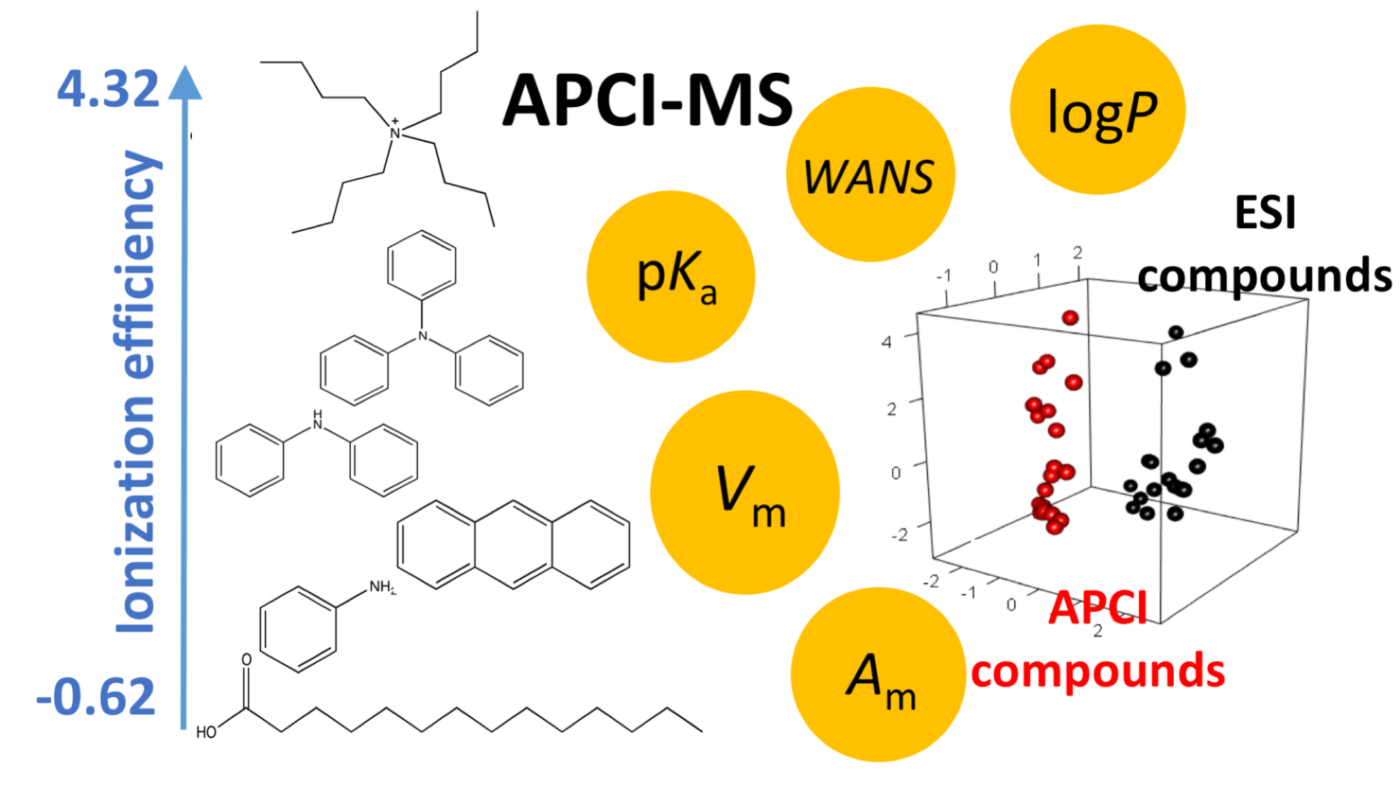
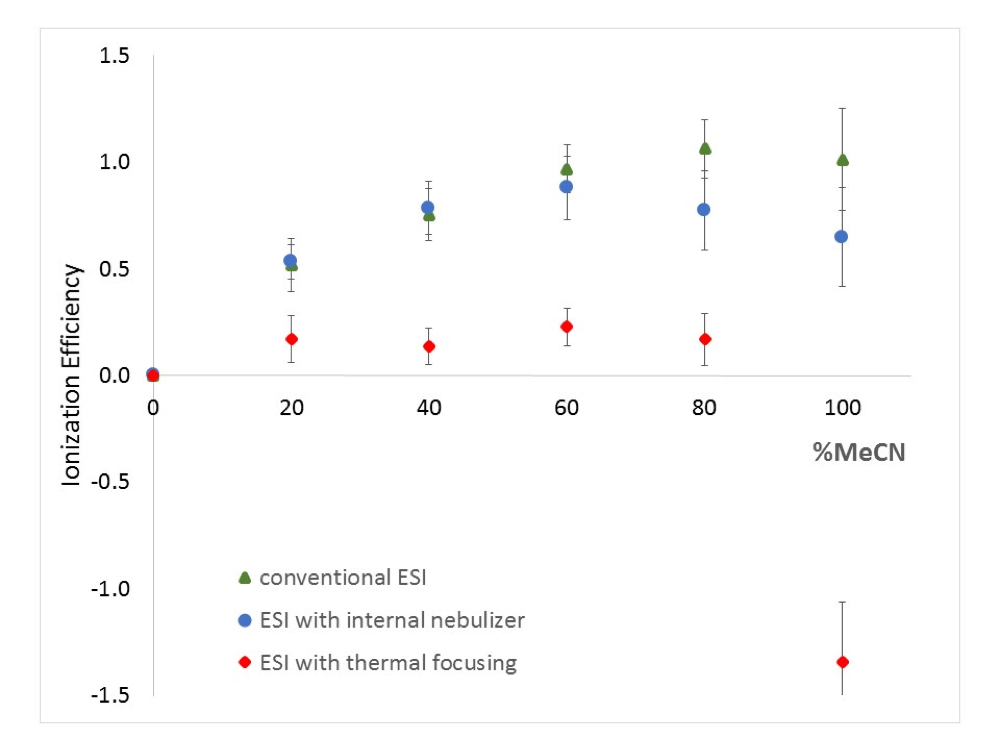
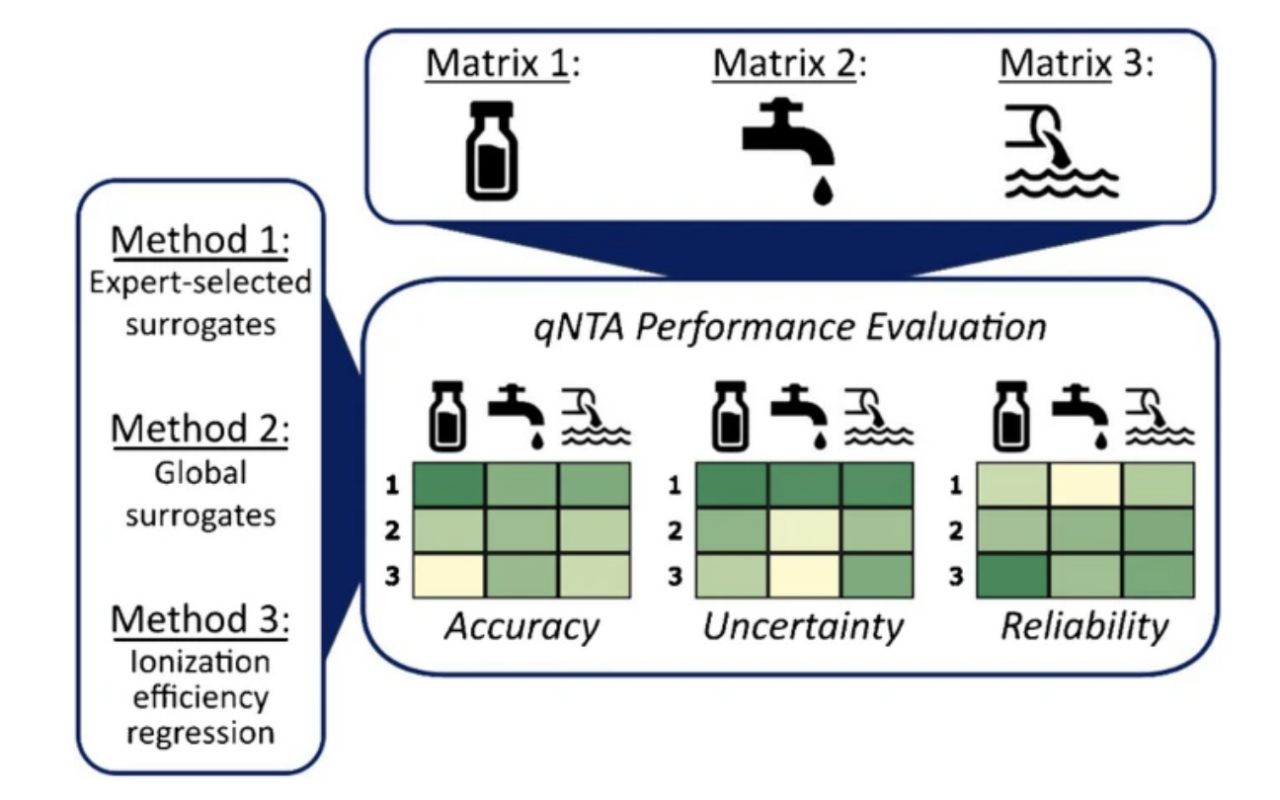
Non-targeted analysis (NTA) is commonly used for the detection and identification of emerging pollutants, including many per- and polyfluoroalkyl substances (PFAS). While NTA outputs are often non-quantitative, concentration estimation is now possible using quantitative non-targeted analysis (qNTA) approaches. To date, few studies have examined matrix effects on qNTA performance, and little is therefore known about the implications of matrix effects on qNTA results and interpretations. Using a set of 19 PFAS, we examined the impacts of drinking water (DW) and waste-activated sludge matrices on qNTA performance across three qNTA approaches: one structure-independent approach based on “global” surrogates and two structure-dependent approaches based on “expert-selected” surrogates and predicted ionization efficiency (IE) regression. The performance of each qNTA approach was examined separately for the PFAS prepared in pure solvent, DW extract, and sludge extract using leave-one-out modeling. Performance was evaluated using previously defined qNTA metrics that describe predictive accuracy, uncertainty, and reliability. The studied sample matrices had minimal effects on qNTA accuracy and larger effects on qNTA uncertainty and reliability. Using solvent-based surrogate data to inform matrix-based estimations yielded lower uncertainty, but also lower reliability, emphasizing that uncertainty must be considered in context of reliability. No single qNTA approach uniformly performed best across all comparisons. Since the IE regression and global surrogates approaches proved most reliable, we recommended them for future qNTA applications. This study highlights the importance of examining multiple performance metrics and utilizing matrix-matched surrogate data in qNTA studies.
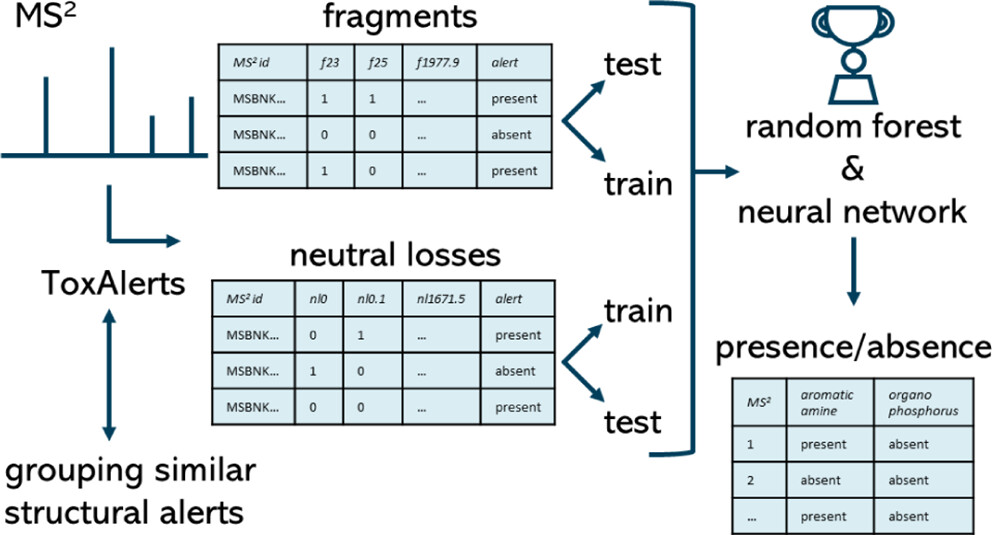
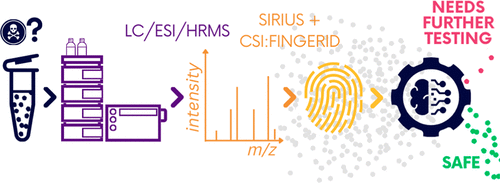
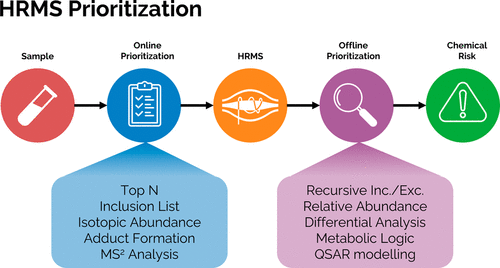
Machine Learning-based Classification for the Prioritization of Potentially Hazardous Chemicals with Structural Alerts in Nontarget Screening
Nienke Meekel, Anneli Kruve, Marja H Lamoree, Frederic M Been
ES&T 2025
Nontarget screening (NTS) with liquid chromatography high-resolution mass spectrometry (LC-HRMS) is commonly used to detect unknown organic micropollutants in the environment. One of the main challenges in NTS is the prioritization of relevant LC-HRMS features. A novel prioritization strategy based on structural alerts to select NTS features that correspond to potentially hazardous chemicals is presented here. This strategy leverages raw tandem mass spectra (MS2) and machine learning models to predict the probability that NTS features correspond to chemicals with structural alerts. The models were trained on fragments and neutral losses from the experimental MS2 data. The feasibility of this approach is evaluated for two groups: aromatic amines and organophosphorus structural alerts. The neural network classification model for organophosphorus structural alerts achieved an Area Under the Curve of the Receiver Operating Characteristics (AUC-ROC) of 0.97 and a true positive rate of 0.65 on the test set. The random forest model for the classification of aromatic amines achieved an AUC-ROC value of 0.82 and a true positive rate of 0.58 on the test set. The models were successfully applied to prioritize LC-HRMS features in surface water samples, showcasing the high potential to develop and implement this approach further.